Electron Transfer in Nanostructrures
Electron transfer (ET) is ubiquitous in molecular and condensed matter systems. It has long been studied in association with electrochemistry, catalysis, photochemistry, solar energy conversion, and photosynthesis, among other topics of interest in physics, chemistry, and biology. As interests evolve to more complex molecular systems consisting of heterogeneous and supramolecular structures in molecular and polymeric photosynthetic systems, the internal (nuclear) degrees of freedom have become relevant, and sometimes crucial. The understanding of the underlying mechanisms responsible of charge and energy transfer in molecular assemblies is important because, on the atomic scale, structural dynamics strongly influences function and electronic behavior. Such computational approaches also provide theoretical support to the experimental counterpart, which consists of ultrafast time-resolved techniques available for the study of charge and energy transfer processes that occur in supra-molecular structures, clusters, quantum dots, and various heterostructures.
The endeavor to simulate the quantum dynamics of energy and charge-transfer processes in large molecular structures and under realistic conditions has only recently been accomplished within the atomistic framework. The DynEMol method consists of a quantum−classical Ehrenfest approach for nonadiabatic excited-state molecular dynamics. The quantum mechanical part of the method is based on the extended Hückel formalism, whereas the nuclei are treated via molecular mechanics (MM). The self-consistent coupling between quantum and classical degrees of freedom is calculated on-the-fly by nonadiabatic Hellmann−Feynman−Pulay forces that conserve the total (quantum−classical) energy. The method combines two simple though efficient computational schemes to describe complex excited state effects.
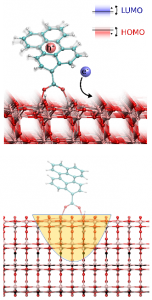
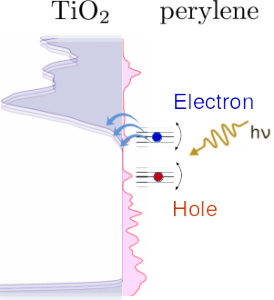
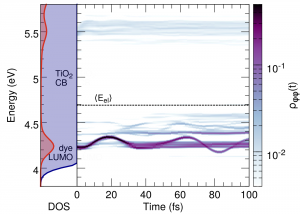
Interfacial Electron Transfer