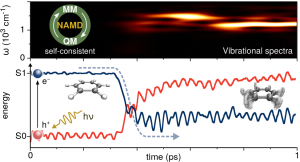
We have developed a mixed self-consistent quantum-classical Ehrenfest approach for nonadiabatic molecular dynamics (NA-MD) simulations in the excited state. The quantum mechanical part of the method is based on the extended Hückel formalism whereas the nuclei are treated with the molecular mechanics method. The self-consistent coupling between quantum and classical degrees of freedom is achieved by nonadiabatic generalized Hellmann-Feynman forces that conserve the overall (quantum-classical) energy. The method combines two simple though efficient computational schemes to describe complex excited state effects such as intramolecular vibronic relaxation and photoisomerization dynamics in atomistic molecular systems.

The semiempirical approach shows good agreement with calculations performed with ab-initio quantum chemistry methods.
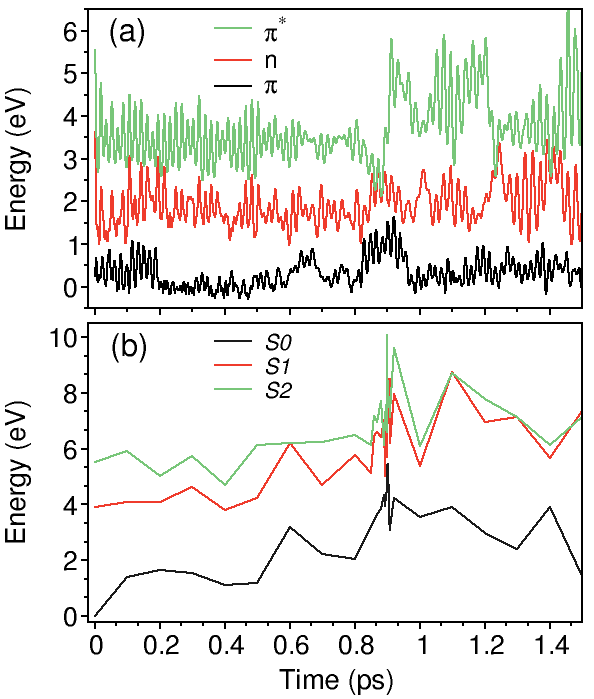
a) HOMO (red) and LUMO (blue) energies during IVR in benzene. The nonradiative decay happens through the S 1 → S 0 conical intersection.
b) Total energy calculations performed with the CASSCF(6,6)/6-31G* method on the NA-MD benzene geometries.
The total energy of the S 0 state (-6280 eV) is set to zero at t=0 for the sake of comparison with a).
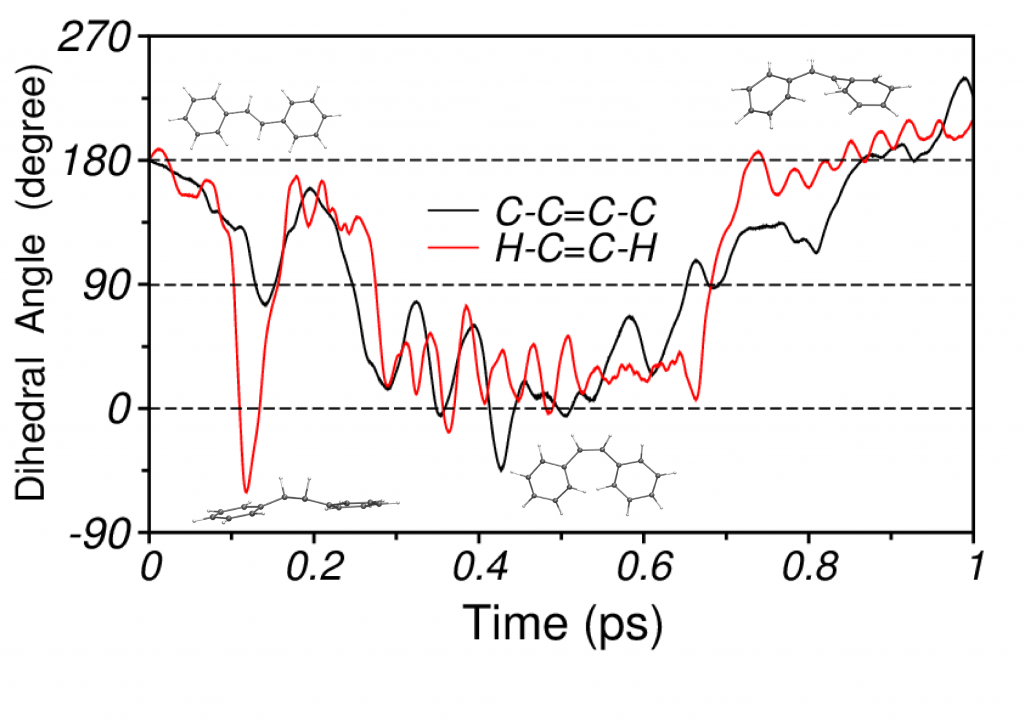
Photoisomerization of Stilbene
For stilbene, our simulations demonstrate that the isomerization mechanism consists of a two step process, which initiates with an abrupt torsion of the H−C=C−H dihedral, triggered by hydrogen out-of-plane (HOOP) vibrations, followed by the rotation of the phenyl rings around the ethylenic bond. The short-time vibrational density of states (VDOS) that is calculated from the excited state NA-MD trajectory shows strong signatures of C−C stretching, C−H rocking and HOOP modes.
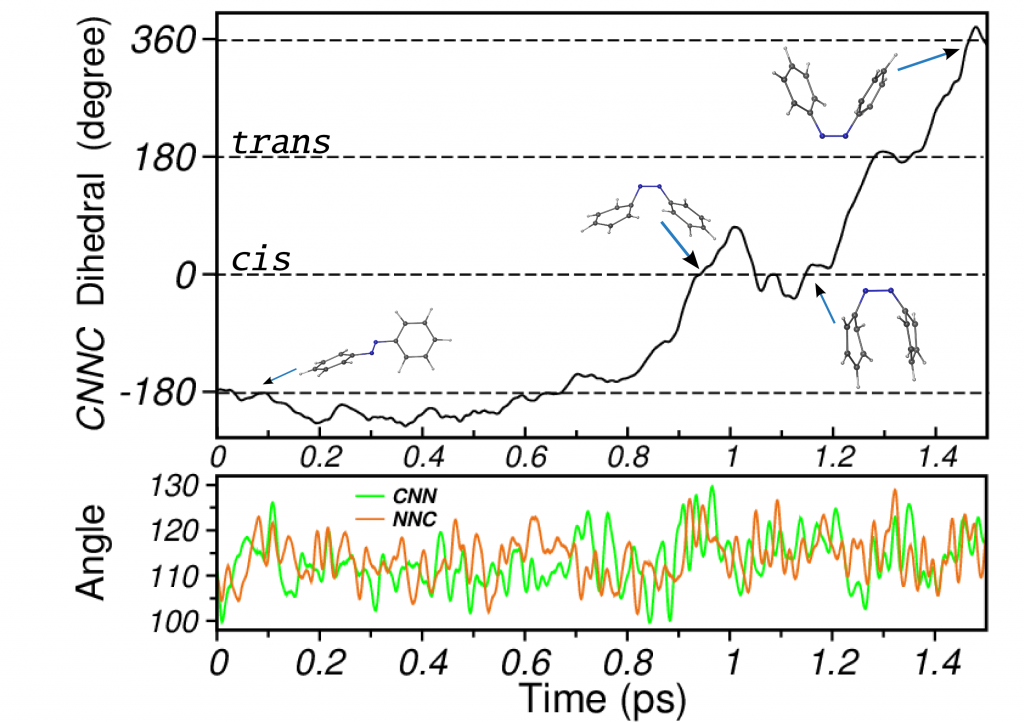
Photoisomerization of Azobenzene
Azobenzene is a prototypical photochromic molecule. Its reversible photoisomerization properties rendered it various applications in photoswitches and nanoscale devices. Azobenzene derivatives are the most used compounds among photoswitches owing to their good light absorption, photoisomerization efficiency and large relative conformational changes. Besides the technological interest, knowledge of the underlying molecular mechanisms driving its photochromism has prompted several studies. Our simulations describe the isomerization mechanism of azobenzene as a multidimensional one, with rotation dominating the process but assisted by inversion (rotation assisted by inversion).
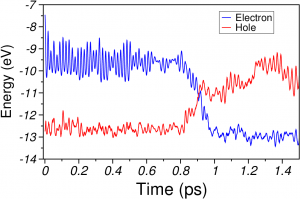
The dynamics calculatations gained from the mixed self-consistent Molecular Mechanics/Extended Hückel Ehrenfest method are supported by ab-initio quantum chemical calculations at the CASSCF level performed for molecular geometries taken from the dynamics calculations.
Nonadiabatic charge transfer processes on large scale molecular systems comprised of photosensitizers and photochemical switches adsorbed on extended solid surfaces are also discussed.
[1] Oliboni, R.S.; Bortolini, G.; Torres, A.; Rego, L.G.C. J. Phys. Chem. C, 2016, 10.1021/acs.jpcc.6b09606
[2] Torres, A.; Oliboni, R.S.; Rego, L.G.C. J. Phys. Chem. Lett. 2015, 6, 4927.